Basic Pharmacology
How Do Drugs Work?
Did you ever wonder how aspirin knows to go to your head when you have a headache and to your elbow when you have “Tennis Elbow”? Or how one or two small aspirins containing only 325-650 mg of active drug can relieve a headache or ease the inflammation of a strained muscle or tendon in a 195 lb. athlete?
The answer to the first question is that drugs are distributed throughout the body by the blood and other fluids of distribution (see distribution below). Once they arrive at the proper site of action, they act by binding to receptors, usually located on the outer membrane of cells, or on enzymes located within the cell.
What Are Receptors?
Receptors are like biological “light switches” which turn on and off when stimulated by a drug which binds to the receptor and activates it. For example, narcotic pain relievers like morphine bind to receptors in the brain that sense pain and decrease the intensity of that perception. Non-narcotic pain relievers like aspirin, Motrin (ibuprofen) or Tylenol (acetaminophen) bind to an enzyme located in cells outside of the brain close to where the pain is localized (e.g., hand, foot, low back, but not in the brain) and decrease the formation of biologically-active substances known as prostaglandins, which cause pain and inflammation. These “peripherally-acting” (act outside of the central nervous system (CNS)) analgesics may also decrease the sensitivity of the local pain nerves causing fewer pain impulses to be sensed and transmitted to the brain for appreciation.
In some instances, a drug’s site of action or “receptor” may actually be something which resides within the body, but is not anatomically a part of the body. For example, when you take an antacid like Tums or Rolaids, the site of action is the acid in the stomach which is chemically neutralized. However, if you take an over-the-counter (OTC) medication which inhibits stomach acid production instead of just neutralizing it (e.g., Tagamet (cimetidine) or Pepsid-AC (famotidine)), these compounds bind to and inhibit recptors in the stomach wall responsible for producing acid.
Another example of drugs which bind to a receptor that is not part of your body are antibiotics. Antibiotics bind to portions of a bacterium that is living in your body and making you sick. Most antibiotics inhibit an enzyme inside the bacteria which causes the bacteria to either stop reproducing or to die from inhibition of a vital biochemical process.
In many instances, the enzyme in the bacteria does not exist in humans, or the human form of the enzyme does not bind the inhibiting drug to the same extent that the bacterial enzyme does, thus providing what pharmacologists call a “Selective Toxicity”. Selective toxicity means that the drug is far more toxic to the sensitive bacteria than it is to humans thus providing sick patients with a benefit that far outweighs any risks of direct toxicity. Of course, this does not mean that certain patients won’t be allergic to certain drugs.
Penicillin is a good example to discuss. Although penicillin inhibits an enzyme found in sensitive bacteria which helps to “build” part of the cell wall around the outside of the bacteria, and this enzymatic process does not occur in human cells, some patients develop an allergy to penicillin (and related cepahlosporin) antibiotics. This allergy is different from a direct toxicity and demonstrates that certain people’s immune system become “sensitized” to some foreign drug molecules (xenobiotics) which are not generally found in the body.
As medical science has learned more about how drugs act, pharmacologists have discovered that the body is full of different types of receptors which respond to many different types of drugs. Some receptors are very selective and specific, while others lack such specificity and respond to several different types of drug molecules.
To date, receptors have been identified for the following common drugs, or neurotransmitters* found in the body: narcotics (morphine), benzodiazepines (Valium, Xanax), acetylcholine* (nicotinic and muscarinic cholinergic receptors), dopamine*, serotonin* (5-hydroxytryptamine; 5-HT), epinephrine (adrenalin) and norepinephrine* (a and b adrenergic receptors), and many others.
Neurotransmitters* are chemicals released from the end of one neuron (nerve cell) which diffuse across the space between neurons called the synaptic cleft and stimulate an adjacent neuron to signal the transmission of information.
The rest of this section is designed to explain the complicated journey of a drug through the body, which pharmacologists call pharmacokinetics.
Basic Pharmacokinetics
Pharmacokinetics is the branch of pharmacology which deals with determining the movement (kinetics) of drugs into and out of the body. Experimentally, this is done by administering the drug to a group of volunteer subjects or patients and obtaining blood and urine specimens for subsequent quantitative (how much) analysis. When the results of these analyses are plotted on graph paper with blood levels or urinary excretion on the verticle axis and time on the horizontal axis, a blood level-time or urinary excretion pattern is obtained.
These graphs can be used to calculate the rates of appearance and elimination of the drug in the bloodstream, the rates of formation of the compounds into which the drugs are transformed in the liver (metabolized), and finally the rates of elimination or excretion of the metabolites.
There are four scientific or pharmacokinetic processes to which every drug is subject in the body:
- ABSORPTION
- DISTRIBUTION
- METABOLISM
- EXCRETION
These four processes occur contemporaneously until (1) all of the drug is absorbed from the GI tract, the muscle or subcutaneous tissue site into which it was injected, and there is no more absorption phase, and (2) all of the drug has been metabolized, and there is no more “parent” drug and it is no longer detectable in the blood.
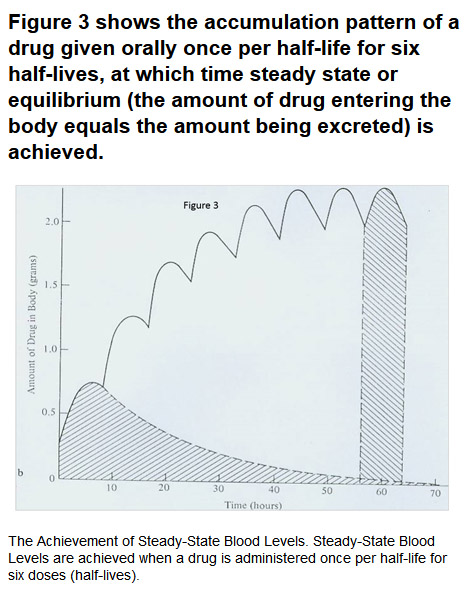
Figure 1 depicts the four contemporaneous pharmacokinetic processes. Figure 2 depicts the blood level-time profile of a single oral and intravenous (IV) dose of a drug. Figure 3, shows the accumulation pattern of a drug given orally once per half-life for six half-lives, at which time steady-state or equilibrium (the amount of drug entering the body equals the amount being excreted) is achieved.
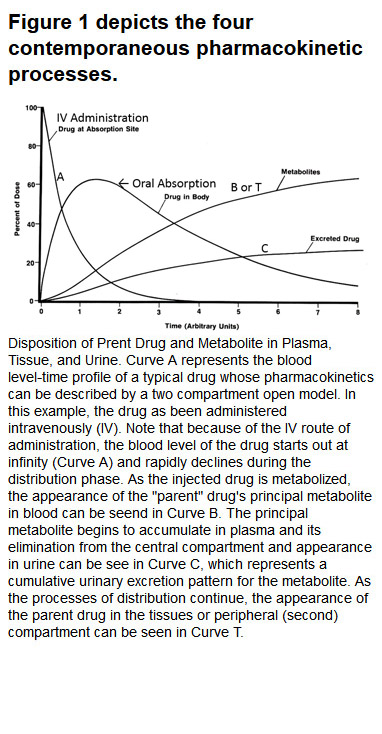{kind=link}
{kind=link}
{kind=link}
Absorption
Absorption is the process by which a drug is made available to the fluids of distribution of the body (e.g., blood, plasma, serum, aqueous humor, lymph, etc.).
In the fasting state, most orally-administered drugs reach a maximum or “peak” blood concentration within 1-2 hours. Intravenous (IV) administration is the most rapid route of administration, with intra-nasal, smoking (inhalation), sublingual (under the tongue), intra-muscular (IM), subcutaneous (e.g., under the skin, SC or SQ), and percutaneous (through the skin) being the next most rapid.
The RATE of absorption of orally-administered drugs and the subsequent appearance of the drug in the blood is dependent on the following factors:
- The rates of disintegration and dissolution of the pill or capsule in the stomach or gastrointestinal (GI) tract,
- The solubility of the drug in stomach or intestinal fluids (the more soluble, the faster),
- The molecular charge on the drug molecule (charged substances are soluble, but don’t pass through lipid (fat) soluble biologic membranes well),
- Aqueous (water) solubility vs. lipid (fat) solubility. Water-soluble drugs are soluble but don’t pass through lipid-soluble biologic membranes well,
- The presence or absence of food in the stomach (food delays the absorption of some drugs and enhances the absorption of others),
- The presence of any concomitant medication(s) that can interfere with gastrointestinal (GI) motility, e.g., Reglan increases GI motility, Aluminum antacids slow, drugs like atropine or scopolamine used for ulcers or “queasy stomachs” slow GI motility keeping some drugs in the stomach slowing absorption, while drugs like Tagamet, Zantac and Prilosec (Pepcid-AC) decrease gastric acid production increasing the rate of gastric emptying and increasing the rate of absorption of some drugs.
Distribution
Once a drug has been absorbed from the stomach and/or intestines (GI Tract) into the blood, it is circulated to some degree to all areas of the body to which there is blood flow. This is the process of distribution. Organs with high blood flow e.g., brain, heart, liver, etc. are the first to accumulate drugs, while connective tissue and lesser perfused organs are the last.
Many drugs are bound to plasma proteins such as albumin. Since only drugs which are not bound are free to exert a pharmacologic effect, the ratio of “free” to “bound” drug is important in determining the onset and duration of action of drugs. Highly bound drugs are distributed less extensively throughout the body and are slower to act. By virtue of their high binding to plasma proteins, they also stay in the body for longer periods of time because the binding sites act as a sort of “reservoir” for the drug, releasing drug molecules slowly.
Metabolism
Drugs in the blood and tissues must be inactivated and excreted from the body. This process is initiated by altering the chemical structure of the drug in such a way as to promote its excretion. The transformation of the drug molecule into a chemically related substance that is more easily excreted from the body is called metabolism, biotransformation or detoxification.
In the case of ethanol, the alcohol molecule is metabolized in the liver by the enzyme alcohol dehydrogenase, to acetaldehyde which causes dilatation of the blood vessels and, after accumulation, is responsible for the subsequent hangover which ensues. The acetaldehyde is subsequently metabolized by the enzyme aldehyde dehydrogenase to acetate, a substance very similar to acetic acid or vinegar.
Therapeutic agents like antibiotics and drugs used for the treatment of high blood pressure, epilepsy (e.g., phenobarbital, Dilantin), pain (e.g., morphine, codeine), anxiety (e.g., Valium, Xanax) are also metabolized to chemically-related compounds called metabolites, which are then excreted in the urine.
REMEMBER: Urine drug screens usually determine metabolites in urine, not the original “parent” drug which was ingested or taken. For example, if cocaine is snorted, smoked or injected, a urine drug screen will most often detect the cocaine metabolite benzoylecgonine in the urine, not cocaine itself. The same analogy applies to other drugs of abuse like: heroin, morphine, amphetamines, PCP (Angel Dust), barbiturates, marijuana, etc.
Excretion
Excretion is the process by which a drug is eliminated from the body.
Drugs can be excreted by various organs including the kidney and lungs, and found in many biological fluids like: bile, sweat, hair, breast milk, or tears. However, the most common fluid in which to look for drugs is the urine.
In order to determine the rate of excretion of any drug from blood, one must first be certain that all the drug in the subject’s GI tract has been absorbed. If not, calculation of a rate of excretion would be confounded be the ongoing absorption of more drug. Once all the drug has been absorbed, this is called the post-absorbtive, or distributive stage. At this time, serial (multiple) blood level determinations should show a decline with time. The slope of the log concentration-time graph is called the half-life (T1/2) and is indicative of the drug’s half-life, or rate of excretion. The half-life represents the amount of time required to eliminate half of the drug from the body.
Figure 4 shows a typical plot of the log of the drug’s blood concentration on the verticle axis vs. time on the horizontal axis. The log of the blood concentration is used to convert the curved portions of the graph shown in Figure 2 to a straight line.
{kind=link}
Generally, it takes six half-lives to rid the body of 98% of a drug and 10 half-lives to completely eliminate the drug from the body. Using these mathematical relatonships allows pharmacologists to determine how often a therapeutic drug should be administered to a patient or toxicologists to determine a time interval within which one would test positive for drugs of abuse. Table I shows the approximate time intervals individuals will test positive in blood and urine for common drugs of abuse.
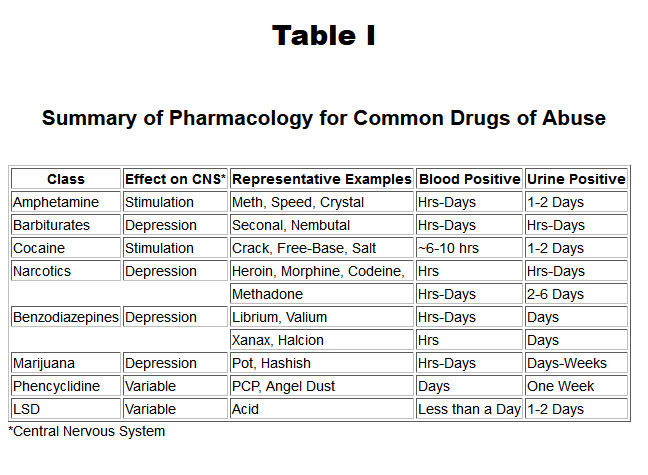{kind=link}
Correlating Drug or Ethanol Blood Levels and Positive Urine Tests With Effects of Impairment
A statutory level for the presumption of DWI is just that, an arbitrary standard. Any BAC level, whether 0.10% or 0.08%, speaks only to a legal standard, and not a scientific (physiological) standard.
The same analogy applies for intoxicating drugs or drugs with an ability to impair an individual’s mental or physical capacity. In order for an individual to be presumed under the influence of an intoxicating or mind-altering drug, it is necessary to establish that there was a significant or pharmacologic concentration of the drug present in the individual’s bloodstream, and that the individual was not sufficiently tolerant to the effects of the drug such as to mitigate any intoxicating effect(s). For example, an individual who had been taking a drug known to cause sedating or intoxicating effects to which the subject could become tolerant, could not be presumed to be too impaired to drive while taking the medication if the subject had been taking the drug long enough to permit the development of tolerance to the sedating or intoxicating properties of that drug.
Some examples would include benzodiazepines like Valium or Xanax, tricyclic antidepressant drugs like Elavil or Tofranil, anti-epileptic drugs like barbiturates and Dilantin which induce their own metabolism and are excreted more rapidly after they have been taken for several weeks, or narcotics like codeine, which are well-known to produce tolerance. In situations like these, it is important to obtain the testimony of other individuals such as co-worders or family members who can corroborate the lack of observable impairment in the subject following drug ingestion.
If an individual is accustomed to having 2-3 (or more) alcoholic drinks per day, with dinner or while watching TV after work, it is quite likely that they will have developed some tolerance to the intoxicating properties of alcohol and might not show signs of intoxication even at BACs over 0.10%. On the other hand, an individual who drinks infrequently would have developed no tolerance and might show signs of intoxication at BACs below the statutory level.